|
O alinhamento local consiste no alinhamento de apenas parte das sequências envolvidas. É feita uma procura por regiões com semelhança local e não é considerada a sequência em todo o seu comprimento. O algoritmo de Smith-Waterman é usado no âmbito do alinhamento local de pares de sequências.
Para exemplifica o funcionamento deste algoritmo são escolhidas duas sequências de aminoácidos:
HEAGAWGHEE (sequência #1)
PAWHEAE (sequência #2)
Temos que M = 10 (comprimento da sequência #1) e N = 7 (comprimento da sequência #2)
Este algoritmo de programação dinâmica é dos mais simples usados para este tipo alinhamento e tal como o algoritmo de Needleman-Wunsch, também se processa em três passos: inicialização, preenchimento da matriz (scoring) e Alinhamento (traceback).
- Inicialização
- O primeiro passo é criar a matriz com M + 1 colunas (uma coluna é para o espaçamento) e N + 1 linhas onde M e N correspondem ao comprimento das sequências a alinhar.
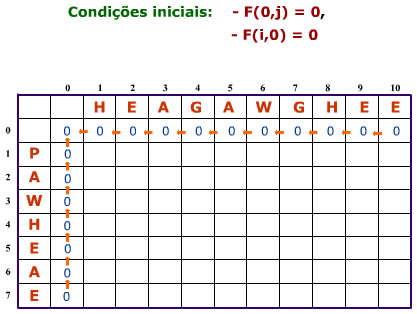
- Inicializar a matriz de acordo com a condição inicial, sendo que F(i,j) é a Função de Pontuação para o preenchimento da matriz e d a pontuação do espaçamento:
- Preenchimento da Matriz (scoring)
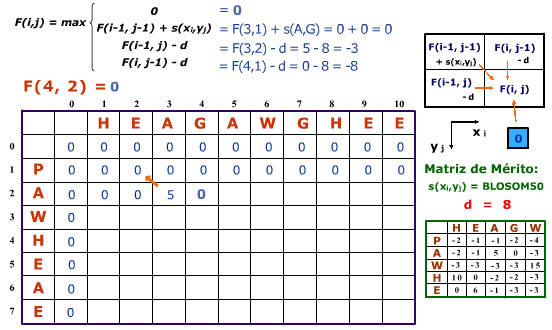
- Escolher a Função de Pontuação e a Matriz de Mérito

- Preencher o resto da tabela de acordo com a função de pontuação e a matriz de mérito.
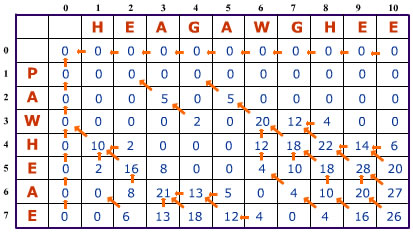
A matriz totalmente preenchida fica da seguinte forma:
- Alinhamento (traceback)
- A pontuação máxima de alinhamento local é 28, que se encontra na posição 9,5.
- Começar na posição com pontuação máxima (9,5 neste caso) e olhar para as células vizinhas que podem ser as precedentes directos de acordo com a seguinte regra de alinhamento:
- Diagonal: xi alinha com yj;
- Cima: yi alinha com espaço;
- Esquerda: xj alinha com espaço;
Na realidade, para o primeiro passo, significa olhar para a diagonal (xi alinha com yj).
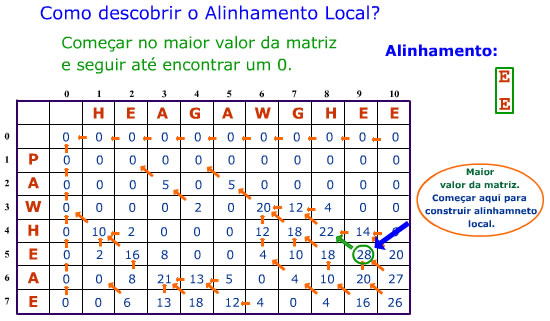
- Seguir todo o caminho até alcançar uma célula com valor 0 (zero).
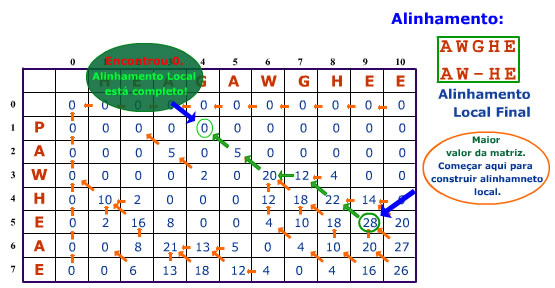
O valor na posição 9,5 é dado pela soma das parcelas correspondentes à pontuação de cada alinhamento individual. Segue-se uma animação, que ajuda compreender o funcionamento deste algoritmo para uma alinhamento entre estas duas sequências de aminoácidos:
|
