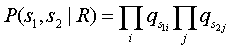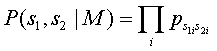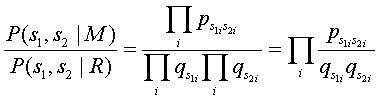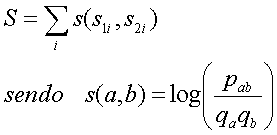|
As matrizes de mérito são também conhecidas por matrizes de substituição. Estas matrizes surgem da necessidade de atribuir um valor ao alinhamento de cada par de caracteres.
Um biólogo com uma grande intuição para proteínas poderia sugerir com realismo um conjunto de 210 valores, correspondentes a todos os possíveis alinhamentos entre pares de aminoácidos. No entanto seria útil arranjar alguma teoria que justificasse os valores que surgissem na matriz. Por exemplo, utilizar um modelo probabilístico é uma possibilidade.
Modelo probabilístico
Antes de introduzir este modelo convém definir alguma notação.
Considere um par de sequências, S1 e S2, com tamanhos n e m, respectivamente. Seja s1i o símbolo na posição i da sequência S1 e s2j o símbolo na posição j da sequência S2. Estes símbolos pertencem a um alfabeto, que contém as bases (A,C,T,G) para sequências de DNA ou os 21 aminoácidos para sequências de proteínas. Os símbolos do alfabeto serão identificados pelas letras minúsculas a e b.
Este modelo probabilístico tem por objectivo atribuir ao alinhamento um valor que corresponda a uma probabilidade das sequências alinhadas estarem relacionadas em oposição a não estarem relacionadas. Este valor é obtido utilizando dois modelos que atribuem uma probabilidade a cada uma das situações referidas, e onde depois é calculado o cociente entre as duas probabilidades.
Para obter as matrizes de substituição, utilizando este modelo considera-se que o alinhamento entre as sequências é global e que não são introduzidos espaçamentos.
Modelo aleatório ou não relacionado, R
Este modelo assume que o caractere a ocorre de uma forma independente com uma probabilidade qa. A probabilidade das duas sequências é apenas o produto das probabilidades de cada caractere:

Modelo relacionado, M
Neste modelo considera-se que os pares de caracteres alinhados ocorrem com uma probabilidade conjunta pab. Esta probabilidade pode ser vista como a probabilidade dos caracteres a e b terem sido gerados de uma forma independente a partir do mesmo caractere original c. A probabilidade atribuída ao alinhamento em análise é determinada pela seguinte expressão:
O cociente entre as probabilidades obtidas pelos modelos M e R, é conhecido por odds ratio:
Por forma a ser possível utilizar estas probabilidades numa função de mérito aditiva, utiliza-se o logaritmo dos valores obtidos pela expressão anterior.
Desta forma a função de mérito seria:
O valor s(a,b) obtido para cada par de caracteres dá uma indicação da importância desse par ocorrer alinhado relativamente a ocorrer desalinhado. Estes valores são normalmente apresentados em forma de matriz, designada de matriz de substituição.
As matrizes PAM (Point Accepted Mutation) foram as primeiras matrizes de substituição de aminoácidos, utilizadas nos alinhamentos efectuados durante uma pesquisa de sequências homólogas em base de dados biológicas.
A escolha da matriz de substituição influência fortemente o resultado dos alinhamento. É normal encontrarem-se citações que sugerem que a escolha da melhor matriz é a tarefa mais crítica numa operação de pesquisa de sequências homólogas numa base de dados.
Idealmente a matriz de substituição deveria reflectir o fenómeno biológico que um alinhamento procura mostrar. Por exemplo, na análise de sequências que divergiram devido a um processo evolutivo com mutações, os números da matriz de substituição deveriam ser obtidos a partir de observações de sequências ancestrais e das respectivas sequências descendentes. Já para a situações em que são analisados motivos conservados, então os números da matriz deveriam ser obtidos observando conjuntos de sequências que contenham esses motivos.
Construção das matrizes PAM
O termo PAM tem 2 utilizações distintas mas relacionadas. Primeiro, pode ser utilizado como unidade de medida da distância evolutiva entre duas sequências de aminoácidos. É possível afirmar-se que a sequência S1 está a uma distância de 3PAM da sequência S2. Na segunda situação o termo PAM é utilizado para referenciar uma determinada matriz de substituição de aminoácidos. No entanto, os valores destas matrizes apresentam uma relação com as unidades PAM.
Estas matrizes foram iniciamente propostas por Margaret Dayhoff e seus colaboradores [Atlas of Protein Sequence and Structure, 5:345-52, 1978].
Definição: Duas sequências S1 e S2 dizem-se a uma distância evolutiva de 1PAM se uma série de mutações pontuais "aceites" (não são consideradas inserções nem apagamentos) levaram à conversão de S1 em S2 com uma média de uma mutação "aceite" em cada 100 aminoácidos.
O termo "aceite" indica que a mutação foi incorporada na proteína e foi passada à geração seguinte. Isto é, ou a mutação não alterou a função da proteína ou a modificação foi benéfica para o organismo, pelo menos não foi letal.
Um erro comum é pensar que duas sequências de proteínas distanciadas de 1PAM apresentam uma diferença de 1 % entre os aminoácidos que as constituem. Este raciocínio não está correcto e deve-se ao facto de uma única posição da sequência poder sofrer mais do que uma mutação.
Por exemplo, duas sequências que estão distanciadas de 100PAM não são diferentes em todas as suas posições. Na realidade, espera-se que sequências que tenham divergido de 200PAM ainda sejam idênticas em 25% das suas posições e que duas sequências distanciadas de 250PAM possam ainda ser distinguíveis de um par de sequências geradas aleatoriamente.
|